
FROGS Core
Galaxy
- Get Started ▾
- Reads processing
- Remove chimera
- Cluster/ASV filters
- Taxonomic affiliation
- Phylogenetic tree building
- ITSx
- Read demultiplexing
- Affiliatio filters
- Affiliation postprocessing
- Abundance normalisation
- Convert Biom file to TSV file
- Convert TSV file to Biom file
- Cluster/ASV report
- Affiliation report
Main tools
Optional tools
CLI
- Get Started
- Reads processing
- Remove chimera
- Cluster/ASV filters
- Taxonomic affiliation
- Phylogenetic tree building
- ITSx
- Read demultiplexing
- Affiliation filters
- Affiliation postprocessing
- Abundance normalisation
- Convert Biom file to TSV file
- Convert TSV file to Biom file
- Cluster/ASV report
- Affiliation report
Main tools
Optional tools
ASV phylogenetic tree
Context
This is a bioinformatics tool used to reconstruct phylogenetic trees from ASV (Amplicon Sequence Variant) sequences.
It allows researchers to infer evolutionary relationships among microbial sequences based on their similarity.
Phylogenetic trees are essential for diversity analyses, community comparisons, and ecological interpretation in microbial studies.
How it does
The program uses a FASTA file of ASV seed sequences and an abundance BIOM file as input.
It constructs a phylogenetic tree, taking into account sequence similarities to infer branching relationships.
It is designed for datasets with fewer than 10,000 sequences to ensure computational efficiency.
Logs and HTML reports are generated to summarize the tree-building process and provide graphical visualization of results.
Configuration: 16S V3V4 Swarm
mkdir FROGS/SWARM/
sbatch -J tree -o LOGS/tree.out -e LOGS/tree.err -c 8 --export=ALL --wrap="module load devel/Miniforge/Miniforge3 && module load bioinfo/FROGS/FROGS-v5.0.2 && tree.py --input-fasta FROGS/SWARM/filters.fasta --input-biom FROGS/SWARM/affiliation.biom --output-tree FROGS/SWARM/phylo_tree.nwk --log-file FROGS/SWARM/tree.log --html FROGS/SWARM/tree_results.html && module unload bioinfo/FROGS/FROGS-v5.0.2"
tree.py --help)
Interpretation: 16S V3V4 Swarm
This report shows that all ASVs are in the tree.
You can use the tree view output to explore the tree.
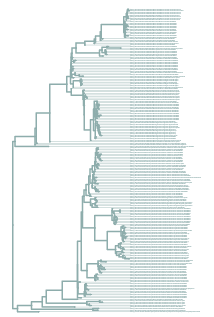
You can use the tree view output to explore the tree.