
FROGS Core
Galaxy
- Get Started ▾
- Reads processing
- Remove chimera
- Cluster/ASV filters
- Taxonomic affiliation
- Phylogenetic tree building
- ITSx
- Read demultiplexing
- Affiliatio filters
- Affiliation postprocessing
- Abundance normalisation
- Convert Biom file to TSV file
- Convert TSV file to Biom file
- Cluster/ASV report
- Affiliation report
Main tools
Optional tools
CLI
- Get Started
- Reads processing
- Remove chimera
- Cluster/ASV filters
- Taxonomic affiliation
- Phylogenetic tree building
- ITSx
- Read demultiplexing
- Affiliation filters
- Affiliation postprocessing
- Abundance normalisation
- Convert Biom file to TSV file
- Convert TSV file to Biom file
- Cluster/ASV report
- Affiliation report
Main tools
Optional tools
Abundance normalisation
Context
This bioinformatics tool is used to normalize microbial abundance data. It ensures that all samples have comparable sequencing depth, which is essential for accurate downstream ecological and diversity analyses. Normalization reduces biases caused by uneven sequencing effort across samples and allows fair comparison of microbial community composition.
How it does
The program takes a BIOM file with abundance data and a corresponding FASTA file with sequences. It randomly samples a fixed number of sequences per sample (--num-reads) or, alternatively, uses the size of the smallest sample (--sampling-by-min) to determine the sampling depth. Samples with fewer sequences than the selected threshold can optionally be deleted (--delete-samples). The process produces a new normalized BIOM file and a corresponding FASTA file containing only the retained sequences. Logs and summary HTML files are also generated for documentation and quality control.
Configuration: 16S V3V4 Swarm
sbatch -J normalisation -o LOGS/normalisation.out -e LOGS/normalisation.err -c 8 --export=ALL --wrap="module load devel/Miniforge/Miniforge3 && module load bioinfo/FROGS/FROGS-v5.0.2 && normalisation.py --input-fasta FROGS/SWARM/filters.fasta --input-biom FROGS/SWARM/filters.biom --output-fasta FROGS/SWARM/normalisation.fasta --log-file FROGS/SWARM/normalisation.log --output-biom FROGS/SWARM/normalisation.biom --html FROGS/SWARM/normalisation.html --sampling-by-min && module unload bioinfo/FROGS/FROGS-v5.0.2"
normalisation.py --help)
Interpretation: 16S V3V4 Swarm
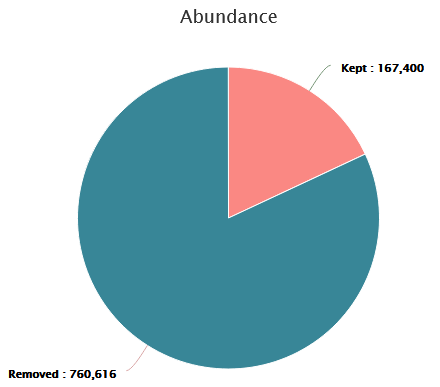
- ~82% of sequences are lost
- 167,400 sequences are remaining