
Still Have Questions?
Start by looking at the “What is FROGS” section to find out what each tool is used for, watch the tutorials to learn how to use them, and check the FAQ for other questions. If you cannot find the answers you're looking for, contact us via support.
General
The HTML report is no longer fully functional!
Problem
HTML reports generated with FROGS versions older than version 5.1.0 can no longer be displayed correctly. A reorganization of the HighCharts library was carried out on their side following a licensing change. As a result, the url used in FROGS HTML reports is no longer valid. It now points to the license-protected version of the library. To resolve the issue, the library path must be updated to the correct location.
Since we've been using Echarts since version 5.1.0, this only applies to files generated with earlier versions.
Solution
You need to modify the web page code to "repair" it. Open the HTML file with a text editor and replace:
with
This must be done for every occurrence (there may be several occurrences per page).
You can automate this process from the command line. For example, if all your HTML reports are stored in the same directory, you can run:
HTML reports generated with FROGS versions older than version 5.1.0 can no longer be displayed correctly. A reorganization of the HighCharts library was carried out on their side following a licensing change. As a result, the url used in FROGS HTML reports is no longer valid. It now points to the license-protected version of the library. To resolve the issue, the library path must be updated to the correct location.
Since we've been using Echarts since version 5.1.0, this only applies to files generated with earlier versions.
Solution
You need to modify the web page code to "repair" it. Open the HTML file with a text editor and replace:
https://code.highcharts.com/8.2.0with
https://cdn.jsdelivr.net/npm/highcharts@8.2.0This must be done for every occurrence (there may be several occurrences per page).
You can automate this process from the command line. For example, if all your HTML reports are stored in the same directory, you can run:
find . -type f -name "*.html" -exec sed -i 's|https://code\.highcharts\.com/8\.2\.0|https://cdn.jsdelivr.net/npm/highcharts@8.2.0|g' {} +Denoising
What should I do if all or almost all of my sequences are eliminated during preprocessing ?
Things to check :
- Review the read quality profiles (quality scores, expected errors) and adjust the thresholds if needed.
- Make sure the minimum and maximum amplicon lengths match your target region.
- Inspect the intermediate FROGS statistics to see at which step the sequences are filtered out.
- Try running a small subset of your data with more permissive parameters to identify which step causes the loss.

How can I find out the expected amplicon sizes (min and max)?
The expected amplicon length depends on your marker gene and the primers used.
You can:
You can:
- Check the literature or reference databases for your target region (e.g., 16S V3–V4 ≈ 400–450 bp, ITS2 ≈ 250–400 bp).
- Align a few reference sequences amplified with your primers to estimate the typical size range.
- If uncertain, start with a broad interval (±20%) and refine it after examining the length distribution in FROGS statistics. For paired reads Illumina only: These parameters correspond to the sizes below/above which assembled paired reads will be discarded. They allow to filter badly assembled paired reads.
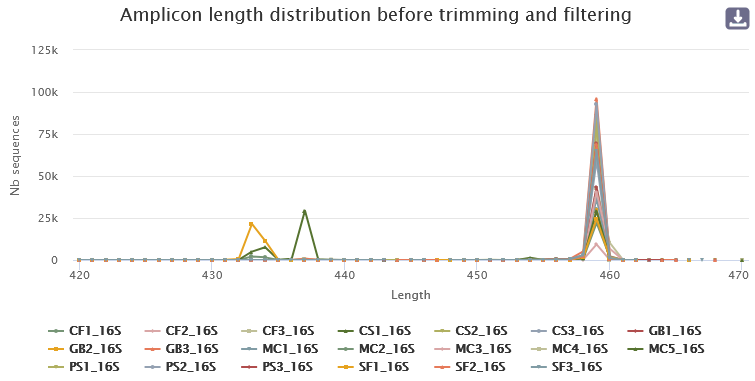
- The length distribution of sequences show that some sequences do not have the expected size. We can run this tool again to increase min amplicon size and reduce max amplicon size.
For example, for this dataset, we first set --min-amplicon-size 200 --max-amplicon-size 490 :
Let look at the HTML file to see the result of denoising.
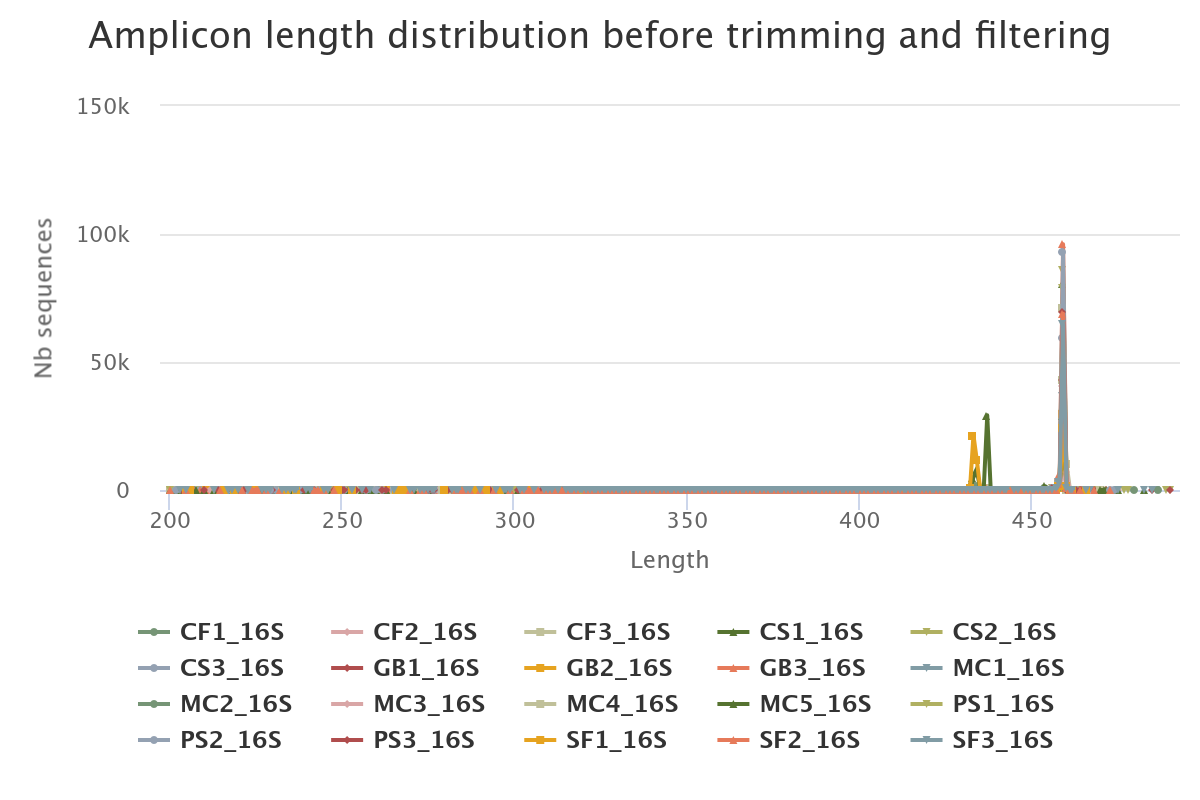
Differences can be seen in the second HTML report.
For long reads, the size of the amplicon can be determined using scientific documentation and/or other tools for studying data quality, such as Fastqc.
How should I correctly enter the sequencing primers?
Is it mandatory to remove the sequencing primers?
When primer sequences are provided, FROGS Core preprocess will automatically remove them from the reads once detected. Choosing not to search for primers when
they are present in the reads involves a risk of generating artifacts: in cases of mismatch between the primer and the target sequence (while still allowing hybridization),
repeated PCR cycles can cause the actual sequence to be progressively overwritten by the primer sequence. This may result in sequencing an artefactual sequence within the
primer region.
Clustering
What is d? Should it be set to 3%?
d is the distance parameter used by the clustering algorithm Swarm. Swarm is a local threshold clustering algorithm and does not rely on any global threshold
(such as the historically used 3%). It iteratively aggregates sequences that differ by d nucleotides: if a path of sequences with d differences between pairs
exists between two reads, they will be grouped into the same cluster. Depending on the actual structure of each cluster, the d parameter results in clusters
with variable percentages of sequence divergence. These percentages adapt to the natural boundaries of each biological entity, without enforcing a fixed threshold.
Therefore, d must not be set to match a desired percentage value.
What value of d should be used?
For short-read sequencing data, it is recommended to use d=1. Given the error rates, read lengths and number of reads, there are typically paths with only one
difference and/or sequencing error between reads that correspond to the same biological entity.
For long-read sequencing data, the error rates, read lengths and read counts generally result in sequences with multiple differences between them, without intermediates differing by only one nucleotide. In such cases, it may be appropriate to test higher d values, keeping in mind that this will significantly increase computational time.
For long-read sequencing data, the error rates, read lengths and read counts generally result in sequences with multiple differences between them, without intermediates differing by only one nucleotide. In such cases, it may be appropriate to test higher d values, keeping in mind that this will significantly increase computational time.
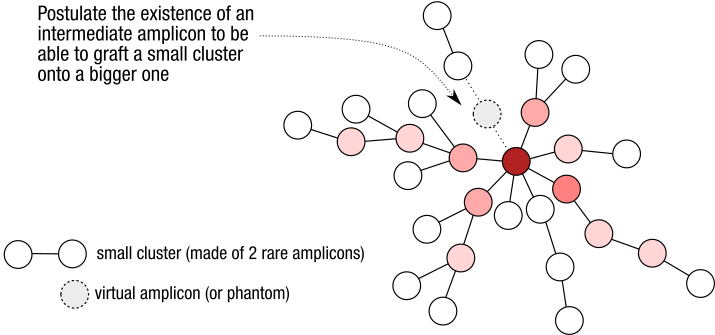
Should you use “refine clustering”? What is it?
The “refine clustering” option, which can only be used with d=1, allows low-abundance clusters to be merged with a larger, more abundant cluster if a connection exists
with d=2. This process helps consolidate biologically related sequences that may have been split due to low coverage or sequencing errors. It is based on the –fastidious
option in Swarm. Swarm github page
Does FROGS produce OTUs or ASVs?
FROGS, when used with Swarm (from version 1 to version 5 and in the future) has always produced local-threshold clusters with properties very similar to ASVs generated by
dada2, before it emerges. Prior to version 4, these cleaned clusters were referred to as OTUs. However, with the rise of ASVs usage, the term “OTU” has increasingly
(and incorrectly) become associated with fixed-threshold clustering and its known limitations.
To avoid the misconception that FROGS could rely on fixed-threshold clustering, the terminology was uptaded in version 4 and FROGS now produces ASVs. This designation is validated by the authors of Swarm, and recognized notably in a recent benchmarking study of metabarcoding pipelines (see Molecular Ecology Resources)
Additionnaly, FROGS 5 allows users to directly integrate dada2 as a replacement or complement to Swarm, enabling the production of strict-sens dada2 ASVs.
To avoid the misconception that FROGS could rely on fixed-threshold clustering, the terminology was uptaded in version 4 and FROGS now produces ASVs. This designation is validated by the authors of Swarm, and recognized notably in a recent benchmarking study of metabarcoding pipelines (see Molecular Ecology Resources)
Additionnaly, FROGS 5 allows users to directly integrate dada2 as a replacement or complement to Swarm, enabling the production of strict-sens dada2 ASVs.
Should I choose swarm or dada2?
In response to increasing demand from our user community, FROGS now includes dada2 as an alternative option to Swarm. Both approaches generally produce similar results
for the reconstruction of high-abundance ASVs, but they may differ when it comes to low-abundance ASVs. At the moment, we do not yet have sufficient long-term feedback
on the use of dada2 within FROGS, and therefore we continue to recommend using Swarm as the default clustering method.
Chimera Detection
What percentages of clusters and chimeric sequences are expected?
The proportion of chimeric sequences in data can vary widely depending on the target region and sequencing techniques. If the target includes internal highly conserved regions (typically when amplifying multiple hypervariable regions of the 16S rRNA gene, such as V1-V3 or V3-V4), it is not uncommon to observe 50-75% of clusters being chimeric. These chimeric clusters are usually low in abundance, which generally corresponds to 5-20% of the total abundance.
For targets less prone to chimera formation, these rates can be significantly lower.
For targets less prone to chimera formation, these rates can be significantly lower.
Cluster Filter
How and why choose filter thresholds with cluster filters?
Applying filters based on cluster abundance and/or occurrence is essential to remove residual sequencing noise (sequences with multiples errors, undetected chimeras, …).
Without this noise removal step, diversity is inevitably overestimated.
The literature on this topic is extensive, but we recommend applying:
The literature on this topic is extensive, but we recommend applying:
- a global abundance filter (meaning a filter based on the sum of a cluster’s abundance across all sample in the dataset) set at 0.005% (Bokulich et al., 2013)
- or an occurrence filter (presence in at least X samples), which can be defined either as a fixed threshold or based on the experimental design (set as a proportion of the number of replicates or pseudo-replicates)
- or both
What percentage of filtered clusters and sequences are expected?
FROGS-swarm applied to short-read sequencing data produces clusters with very high resolution, but also generates a large proportion of low-abundance clusters.
Applying one or both of the recommended filters typically removes a substantial number of clusters, while having only a modest impact on total read abundances:
for high-quality data, more than 90% of clusters may be filtered out, representing only 5-20% of the total abundance.
Long-read data can currently be more challenging, as their length, error rate and sequencing depth often result in poor or no clustering. In such cases, direct taxonomic affiliation approaches may be required (see below)
Long-read data can currently be more challenging, as their length, error rate and sequencing depth often result in poor or no clustering. In such cases, direct taxonomic affiliation approaches may be required (see below)
Long read: What to do if cluster filter eliminates all clusters?
With long-read data, clustering may fail to group reads into clusters, especially when using sequencers with high error rates. In such cases, applying FROGS Core Cluster
filters with standard settings can result in the elimination of all sequences. If adjusting clustering parameters does not solve the issue, currently the only viable
approach is to perform direct taxonomic affiliation on individual reads. However, without effective clustering, this step can be extremely time-consuming, and biological
entities that are distant from those represented in the reference database may be poorly reconstructed, resulting in inaccurate abundance estimates.
Taxonomic Affiliation
Which database should be selected for taxonomic affiliation? (see LEAP)
FROGS offers a wide range of databases for taxonomic assignment. You should select one that matches your target region (16S, ITS, rpoB…). Then, choose between generalist databases and more specialized ones depending on your research topic, subject and goals.
To compare the capabilities and specificities of different databases, FROGS recommends using LEAP.
To compare the capabilities and specificities of different databases, FROGS recommends using LEAP.